Qu’est-ce que les bactéries Gram négatif ?

Ces dernières années, une grande attention a été accordée à l’utilisation des antimicrobiens en médecine vétérinaire. L’utilisation responsable des antibiotiques est essentielle au fonctionnement optimal du médicament, en limitant la résistance et en assurant la sécurité alimentaire. En 2014, Dopharma a publié plusieurs fois dans son bulletin d’information un article contenant des informations pratiques sur les antibiotiques. Ces articles sont fusionnés en un seul document, qui est également disponible pour vous entéléchargement (pdf, anglais), mais vous pouvez également lire le texte ci-dessous.
L’ utilisation responsable des antibiotiques signifie non seulement une réduction de l’utilisation des antibiotiques, mais aussi le choix du bon remède dans une situation particulière. Les outils importants qui peuvent être utilisés pour ce choix sont l’antibiogramme et les formuaires pour les différentes espèces d’animaux. Dans un certain choix en formalités (1er, 2ème, 3ème choix) , en fonction de la sensibilité, peut souvent encore être sélectionné entre différentes substances actives. La connaissance de la pharmacodynamique et de la pharmacocinétique est nécessaire pour arriver à un choix justifié. Même lorsqu’un médicament doit être utilisé hors étiquette dans le cadre des bonnes pratiques vétérinaires (GVP), cette connaissance est essentielle à la justification.
A lire aussi : Les conséquences du stress sur la santé et les solutions pour y faire face
Plan de l'article
Intro
Pharmacodynamique et pharmacocinétique
La pharmacodynamique décrit comment un médicament vétérinaire obtient un effet dans l’organisme. Dans le tableau récapitulatif, les antibiotiques sont classés en fonction du mécanisme d’action. Dans les colonnes, les propriétés principales sont discutées. En plus de ces propriétés, le spectre est également important, mais cela n’est pas pris en compte ici. La pharmacocinétique décrit la façon dont le corps traite un médicament vétérinaire. En cinétique, nous avons quatre composantes importantes : Absorption, Distribution, Métabolisme et Elimination (AMDE).
Le Les principaux paramètres sont : Cmax (concentration maximale), Tmax (temps où la concentration maximale est atteinte), CL (clairance), T1/2EL (demi-vie d’élimination) et Vd (volume de distribution). Ces paramètres dépendent des caractéristiques du médicament vétérinaire, ainsi que de la formulation, de la voie d’administration et de la prescription posologique.
A découvrir également : Les bénéfices surprenants de l'activité physique pour maintenir sa santé
Bactéricide ou bactériostatique
La différence entre les antibiotiques bactéricides et bactériostatiques est bien connue ; les antibiotiques bactéricides tuent les bactéries, les antibiotiques bactériostatiques inhibent la croissance. In vitro, la distinction est difficile à établir ; tous les antibiotiques bactéricides ne tuent pas toutes les bactéries dans le délai imparti, tandis que certains antibiotiques bactériostatiques à des doses suffisamment élevées se révèlent également capables de tuer certaines bactéries. Pratiquement, on croit souvent que les antibiotiques bactéricides sont nécessaires dans les infections aiguës ou lorsque le système immunitaire ne fonctionne pas de manière optimale. Dans Cependant, certaines situations antibiotiques bactériostatiques peuvent être préférables. La mortalité rapide due aux antibiotiques bactéricides peut, par exemple, augmenter les symptômes d’une maladie chez les bactéries contenant des endotoxines.
Constante de lipophilité et de dissociation
Pour être efficace, l’antibiotique doit naturellement atteindre les bactéries dans divers tissus, et il est souvent nécessaire que le médicament puisse passer à travers les membranes cellulaires. En plus du transport actif et de la diffusion à travers les pores, la diffusion passive des médicaments est particulièrement importante pour leur passage à travers les membranes. Pour cette diffusion passive, la constante de lipophilité et de dissociation de la drogue sont particulièrement importantes. La lipophilité est indiquée dans le tableau pour chaque groupe antibiotique. Une substance lipophile élevée peut passer plus facilement la structure phospholipidique des membranes cellulaires qu’une couche de substance lipophile. En conséquence, les substances lipophiles élevées atteignent souvent des concentrations efficaces dans la synovia, l’œil et le liquide céphalo-rachidien. La diffusion de substances modérément lipophiles dans ces tissus dépend de la liaison des protéines plasmatiques, et donc du volume de distribution. En outre, cela est montré dans le tableau.
La constante de dissociation (pKa) d’un médicament indique dans quelle mesure il est ionisé et non ionisé. Le passage d’une membrane cellulaire par diffusion passive n’est possible que si la substance est à l’état syndiqué. S’il y a une différence de pH entre deux tissus (p. ex. le sang et le tissu pulmonaire), un antibiotique s’accumule dans l’un de ces tissus selon le pKa. Les bases faibles s’accumuleront dans le tissu avec le pH inférieur et les acides faibles s’accumuleront dans le tissu avec le pH plus élevé. Ce phénomène est appelé piégeage ionique. Dans le corps, il s’agit principalement de la différence de pH entre le plasma et les tissus et de la différence entre le pH intracellulaire et le pH extracellulaire. Exemples de tissus avec un pH inférieur au pH du plasma sont les poumons, la prostate, le lait et l’urine des carnivores. L’urine des herbivores a un pH plus élevé que le pH du plasma. À titre d’illustration : Pour obtenir des concentrations tissulaires élevées dans les poumons, il est préférable de choisir un antibiotique lipophile avec un pKa élevé.
Concentration ou effet dépendant du temps
Avec les antibiotiques dépendants de la concentration, une augmentation de la concentration assure une inhibition ou une élimination plus rapides et plus efficaces des bactéries.
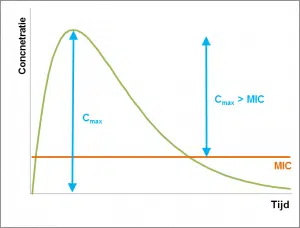
Le paramètre le plus important pour ces antibiotiques est la Cmax > CMI. Il s’agit de la différence entre la concentration plasmatique maximale (Cmax) et la concentration inhibitrice minimale (CMI). Pour obtenir les meilleurs résultats, la Cmax doit être dix fois plus élevée que le MIC. La concentration la plus élevée possible peut être atteinte, entre autres, en rendant les antibiotiques disponibles non pas tout au long de la journée, mais, par exemple, au moyen de dosage du pouls. La durée pendant laquelle la concentration de l’antibiotique reste élevée est moins importante dans ce cas, car ces antibiotiques ont généralement un effet post-antibiotique long (PAE).
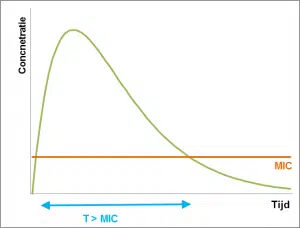
Pour le deuxième groupe d’antibiotiques, l’action dépend de la période pendant laquelle la bactérie est exposée à l’antibiotique. Le paramètre principal est la période au cours de laquelle la concentration est supérieure à la CMI (T> MIC). Pour un effet optimal, la période de concentration au-dessus de la CMI doit être au moins égale à la moitié de l’intervalle posologique. Par exemple, si des études cinétiques montrent que la concentration d’un antibiotique donné reste supérieure à la CMI pendant une période de 6 heures, un intervalle de dose pouvant aller jusqu’à 12 heures doit être utilisé. En général, on peut supposer que ces antibiotiques doivent être offerts aussi souvent que possible pendant la journée, de sorte que le traitement est effectué de préférence en continu.
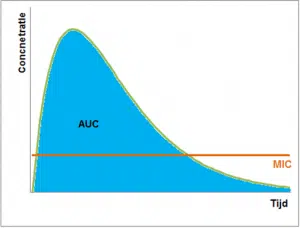
Enfin, il y a encore quelques antibiotiques qui ont les deux propriétés. Une concentration élevée doit donc être atteinte pour ces antibiotiques, mais elle doit également être maintenue. À cette fin, l’ASC (Area Under the Curve) est prise en compte. La valeur par défaut est que l’ASC/CMI doit être supérieure à 125.
Pénicillines
Pharmacodynamique
Le mécanisme bactéricide d’action des pénicillines est basé sur l’inhibition de la synthèse de la paroi cellulaire. Le peptidoglycane formant la paroi cellulaire est composé de plusieurs chaînes peptidiques et de sucres, qui sont reliés entre eux par, entre autres, des protéines de liaison à la pénicilline (PBP) telles que la transpeptidase. Les pénicillines se lient de façon irréversible à ces PBP, de sorte qu’aucun peptidoglycane ne peut être formé. Cet effet ne s’applique qu’à la division des bactéries, car seulement alors la paroi cellulaire est ouverte et le peptidoglycane est nécessaire pour la fermer.
À l’intérieur des pénicillines, il y a une séparation claire entre les pénicillines qui sont contre grammes de bactéries positives et de pénicillines qui ont un large spectre. Cela s’explique en partie par la structure des bactéries. En grammes de bactéries positives, la paroi cellulaire peptidoglycane est située à l’extérieur de la cellule, de sorte que les PBP sont toujours accessibles. En grammes de bactéries négatives, la paroi cellulaire peptidoglycane est encore entourée d’une couche lipidique, ce qui rend les PBP plus difficiles à atteindre. Ceci est illustré dans la figure ci-dessous. Les pénicillines à spectre étroit sont incapables de passer la couche lipidique de grammes de bactéries négatives, tandis que les pénicillines à large spectre le font généralement. D’autres facteurs qui peuvent affecter la sensibilité aux pénicillines comprennent la structure des PBP, la quantité de peptidoglycane (grammes de bactéries positives ont beaucoup plus de peptidoglycane) et la résistance due à la formation de diverses enzymes β-lactamase.
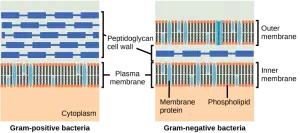
Parce que les pénicillines augmentent la synthèse de la paroi cellulaire , ils ne sont pas efficaces contre les bactéries qui n’ont pas de paroi cellulaire et n’ont donc pas de PBP, comme Mycoplasma spp. Les animaux n’ont pas non plus de PBP, ce qui explique la forte marge de sécurité des pénicillines.
Constante de lipophilité et de dissociation
Les pénicillines ont une faible lipophilité et un petit volume de distribution. Par conséquent, la biodisponibilité des pénicillines n’est généralement pas très bonne. L’exception à ceci est la pénicilline V (phénoxyméthylpénicilline). Cette pénicilline résistante à l’acide a une meilleure biodisponibilité orale que les autres pénicillines. Le faible pKa des pénicillines est responsable du faible volume de distribution ; dans le plasma, la majorité de ces antibiotiques se produisent sous forme ionisée et les membranes sont donc difficiles à passer. Cela conduit au fait que, par exemple, la concentration dans la mamelle est d’environ un cinquième de la concentration plasmatique. Cependant, le passage sur les membranes biologiques améliore quand il y a une inflammation. Cela est dû au fait que le pH des tissus enflammés diminue, à la suite de quoi une plus grande partie des pénicillines sera sous la forme ionisée. Pour cette raison, les pénicillines pendant l’inflammation atteignent encore des niveaux efficaces dans les tissus où ils pénètrent normalement à peine dans.
Concentration ou effet dépendant du temps
Les pénicillines sont des antibiotiques dépendants du temps. Cela signifie qu’ils sont particulièrement efficaces si la bactérie est exposée à l’antibiotique pendant une période suffisamment longue. Le dosage continu est donc préférable au dosage par impulsion. Les préparations injectables sont souvent administrées plusieurs fois par jour ou contiennent des pénicillines liées à la procaïne, ce qui la fait absorber lentement à partir du site d’injection.
En outre, pour les antibiotiques β-lactamines, il existe un phénomène connu sous le nom d’ « effet aigle ». Une concentration élevée apparaît dans certaines bactéries (en particulier les entérocoques spp.) moins efficace que la concentration habituelle. Ceci est causé par l’inhibition de la croissance, ce qui est important pour l’efficacité. Cela souligne l’importance d’un schéma posologique correct avec des concentrations prolongées supérieures à la CMI, mais sans concentrations maximales élevées.
Combinaison d’antibiotiques
Comme décrit précédemment, les pénicillines ne sont efficaces que si elles sont utilisées contre les bactéries divisées. Cela signifie que ces antibiotiques ne peuvent pas être combinés avec des antibiotiques bactériostatiques comme la plupart des inhibiteurs de la synthèse des protéines. Une exception à cela sont les aminoglycosides ; ces antibiotiques sont bactéricides et la combinaison des antibiotiques β-lactamines avec des aminoglycosides agit en synergie parce que les pénicillines augmentent la perméabilité de la paroi cellulaire, ce qui bénéficie aux aminoglycosides.
Résistance
La résistance aux pénicillines est généralement basée sur les β-lactamases. Ces enzymes coupent la β-lactame anneau de pénicillines ouvertes, ce qui les empêche de se lier aux PBP et donc inefficace. Ce mécanisme de résistance est transmis, en particulier, à travers les plasmides. La transmission par plasmides est particulièrement efficace en grammes de bactéries négatives, ce qui entraîne une plus grande variété de β-lactamases chez ces bactéries que dans les grammes de bactéries positives. Cela signifie que le degré d’induction de la résistance aux pénicillines à spectre étroit, qui ne sont efficaces que contre des grammes de bactéries positives, est très faible. D’autre part, les pénicillines à large spectre peuvent induire une résistance relativement rapide à des grammes de bactéries négatives. Le GD Monitoring (2012) montre également que la résistance à ce groupe d’antibiotiques se produit principalement en grammes de bactéries négatives. S.aureus est l’un des rares grammes de bactéries positives où la résistance aux antibiotiques aux pénicillines est un problème. Certaines pénicillines ne sont pas sensibles aux β-lactamases. Un exemple de β-lactamase pénicilline sensible, qui est utilisé vétérinaire est la cloxacilline. En outre, les pénicillines peuvent être combinées avec des inhibiteurs des β-lactamases tels que l’acide clavulanique pour supprimer cette forme de résistance.
D’ autres mécanismes de résistance susceptibles d’intéresser les pénicillines comprennent l’ajustement des PBP et la réduction de la concentration intracellulaire de l’antibiotique par une pompe à efflux ou une réduction de la perméabilité. La réduction de la production de pores normalement utilisés pour passer à travers la membrane de la cellule externe n’est importante que pour les grammes de bactéries négatives telles que E. coli. Ce mécanisme est souvent lié au mécanisme de résistance qui augmente l’efflux d’antibiotiques β-lactamines sur la membrane externe, ce qui n’a de sens que pour les grammes de bactéries négatives.
Les groupes spécifiques de bactéries qui présentent une résistance aux pénicillines sont le SARM, l’ESBL et l’AMPC produisant des germes. Méthicilline Staphylococcus aureus résistant (SARM) est réfractaire à presque tous les antibiotiques β-lactamines. Cette résistance est causée par une mutation chromosomique sur le gène mECA. Ce gène code pour un PBP dont la structure est anormale, ce qui le rend pratiquement insensible aux antibiotiques β-lactamines. Les bactéries productrices de β-lactamase étendue (ESBL) sont réfractaires aux pénicillines, aux céphalosporines et aux monobactammes, car elles décomposent ces antibiotiques β-lactamines. Ces bactéries sont sensibles aux carbapénèmes et aux inhibiteurs de la β-lactamase. Les bactéries produisant de l’AMPC sont un sous-groupe au sein de la bactérie produisant de l’ESBL. Les bactéries qui produisent cette enzyme, contrairement au groupe précédent, sont également insensibles aux inhibiteurs de la β-lactamase. Les carbapénèmes sont alors la seule option de traitement. Il n’y a pas d’antibiotiques homologués vétérinaires dans ce groupe.
Céphalosporines, polymyxines et fluoroquinolones
Pharmacodynamique
Céphalosporines et polymyxines saisir sur la paroi cellulaire des bactéries et les deux ont un effet bactéricide. Les céphalosporines, comme les pénicillines, sont des antibiotiques β-lactamines et inhibe la synthèse de la paroi cellulaire en se liant aux protéines liant la pénicilline (PBP). Pour cette raison, les molécules peptidoglycanes qui composent la paroi cellulaire ne peuvent pas être reliées entre elles. Ce mécanisme d’action est expliqué plus en détail dans le paragraphe sur les pénicillines. Les céphalosporines de première et deuxième génération (céphalexine, céphalonium, cefapirine) ont un spectre étroit et ne sont efficaces que contre les grammes de bactéries positives. Les céphalosporines plus récentes (3ème génération : cefaperazone, ceftiofur ; 4ème génération : cefquinome) sont de plus en plus efficaces contre les grammes de bactéries négatives.
Les polymyxines perturbent la rigidité de la paroi cellulaire des grammes de bactéries négatives en se liant aux lipopolysaccharides (LPS) qui se produisent dans la paroi cellulaire des grammes de bactéries négatives. Cela conduit à réduit l’intégrité de la paroi cellulaire et finalement la mort de la bactérie. LPS est également une endotoxine et la liaison des polymyxines la neutralise également.
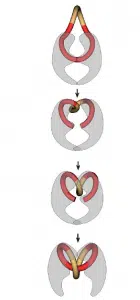
Le calcium et le magnésium se lient également au LPS et la présence de ces ions dans le tractus gastro-intestinal donc, par la compétition, réduit l’efficacité des polymyxines. Les fluoroquinolones inhibent la synthèse bactérienne de l’ADN en empêchant la transcription et la traduction de l’ADN. Cela, ils inhibent les enzymes essentielles de l’ADN gyrase et de la topoisomérase. Ces enzymes garantissent que l’ADN produit dans les bactéries multiplicatrices peut être enroulé. Pour cela, l’ADN est lié à l’une de ces enzymes, coupé ouvert, tordu et fermé à nouveau. Ceci est illustré à la figure 1. Les fluoroquinolones inhibent ces enzymes en formant un complexe avec le brin d’ADN et l’enzyme. En plus d’inhiber la synthèse de l’ADN et restauration de l’ADN, cela assure également la libération de brins d’ADN cassés. Cela crée à nouveau un stress oxydatif pour les bactéries. La combinaison de ces facteurs fait que les fluoroquinolones ont un mécanisme d’action bactéricide.
Le spectre des fluoroquinolones diffère par substance active en raison d’une différence d’affinité pour les enzymes bactériennes. Par exemple, la fluméquine n’est efficace que contre les entérobactéries telles que E. coli et Salmonella spp. En ajoutant une molécule de fluorure, les fluoroquinolones telles que l’enrofloxacine peuvent également se lier aux enzymes de grammes de bactéries positives et d’anaérobies. Cela élargit le spectre et peut également traiter les infections causées par Bordertella bronchiseptica, Manheima haemolytica, Pasteurella spp., Chlamydophila spp. et Actinobacillus pleuropneumoniae.
Constante de lipophilité et de dissociation
Les céphalosporines et les polymyxines ont une faible lipophilité et une petite volume de distribution. Cela entraîne une mauvaise absorption du tractus gastro-intestinal. Après administration parentérale, les céphalosporines et les polymyxines ont une bonne biodisponibilité et sont rapidement absorbées à partir du site d’injection. Cependant, le passage de la membrane reste limité. Ce qui précède explique pourquoi la colistine, en plus de l’administration parentérale, est principalement utilisée comme traitement des troubles gastro-intestinaux. La polymyxine B, à son tour, est principalement utilisée pour le traitement topique des infections. Les céphalosporines sont administrées par voie parentérale ou intramammaire.
Les céphalosporines ont un faible pKa et peuvent donc s’accumuler dans les tissus dont le pH est supérieur au pH du plasma. Les polymyxines ont un pKa élevé, ce qui entraîne une accumulation dans les tissus avec un pH inférieur au pH du plasma.
La fluméquine et les molécules similaires ont une assez bonne biodisponibilité. La nouvelle génération de fluoroquinolones, y compris l’enrofloxacine, cependant, ont une très bonne disponibilité orale. En raison de la lipophilité élevée, du grand volume de distribution et de la faible liaison aux protéines plasmatiques, ces fluoroquinolones plus récentes atteignent également des concentrations tissulaires élevées dans le cerveau, les intestins, le foie et les voies urinaires, entre autres. Les fluoroquinolones s’accumulent dans les cellules phagocytaires, à l’intérieur desquelles elles sont également efficaces contre les bactéries intracellulaires.
La nourriture ralentit l’absorption des fluoroquinolones du tractus gastro-intestinal en raison de la formation complexe avec des ions. Cependant, l’aire sous la courbe (ASC) et donc la biodisponibilité ne changent pas significativement. À des concentrations élevées de magnésium ou d’aluminium dans l’alimentation, la biodisponibilité des fluoroquinolones diminue considérablement.
Les fluoroquinolones sont des amphotères. Cela signifie qu’ils ont deux PKa : un PKa faible et un élevé. En conséquence, ils se répartiront bien aux deux tissus avec un plus haut et aux tissus avec un pH inférieur au pH de la plasma.
Concentration ou effet dépendant du temps
Les céphalosporines ont un effet dépendant du temps. Ainsi, la bactérie doit être mise en contact avec l’antibiotique pendant une période suffisamment longue. Pour un effet optimal, l’intervalle de dosage est déterminé de sorte que cet intervalle ne dépasse pas deux fois la période pendant laquelle la concentration tissulaire est supérieure à la CMI. Cette période devrait être déterminée par espèce sur la base d’études pharmacocinétiques.
Les polymyxines et les fluoroquinolones ont un effet dépendant de la concentration. Avec ces antibiotiques, la durée d’exposition est moins importante que l’obtention d’une concentration élevée sur le site de l’infection. De préférence, une concentration maximale 10 fois plus élevée que la CMI est atteinte.
Combinaison d’antibiotiques
Comme les céphalosporines ne sont efficaces que pour diviser les bactéries, les céphalosporines, ainsi que les pénicillines, ne peuvent pas être combinées avec des bactéries bactériostatiques antibiotiques. L’exception à cette règle est la combinaison avec des aminoglycosides, qui est synergique. Cela a déjà été expliqué dans le paragraphe sur les pénicillines.
Les polymyxines peuvent être combinées avec les sulfamides et le triméthoprime pour un effet synergique contre diverses entérobactéries, y compris P.aeruginosa. Les polymyxines dans la pratique sont souvent combinées avec de l’amoxicilline. Cette combinaison a un effet synergique dans le traitement des infections causées par une combinaison de bactéries. Cette combinaison est également utilisée pour ralentir le développement de la résistance à ces antibiotiques. Humainement, cette combinaison est également utilisée pour traiter les bactéries multirésistantes, y compris P.aeruginosa, E.coli et Enterobacter.
Les fluoroquinolones ont un effet synergique lorsqu’ils sont combinés avec des antibiotiques β-lactamines ou des aminoglycosides.
Résistance
Les principaux mécanismes sur lesquels la résistance contre les céphalosporines est basée sur la modification des PBP, la réduction de la perméabilité de la paroi cellulaire, l’augmentation de l’efflux et l’inactivation enzymatique par les β-lactamases. Ce dernier mécanisme, ainsi qu’avec les pénicillines, est le plus important. Plus d’informations sur la résistance aux antibiotiques β-lactamines peuvent être lues dans le paragraphe sur les pénicillines. Il convient de noter que les nouvelles générations de céphalosporines sont moins sensibles aux β-lactamases que les pénicillines et les céphalosporines de première génération. La différence de sensibilité s’explique par un changement dans la structure en raison de laquelle l’anneau β-lactame est situé à un endroit différent dans la molécule. Aux Pays-Bas, la résistance aux céphalosporines est particulièrement observée en grammes d’agents positifs de mammtite (S.aureus, coagulase négatif staphylocoques) chez le bovin ou b.bronchiseptica chez le porc.
La résistance aux polymyxines n’est guère fréquente. Si la résistance se produit est basé sur une réduction du nombre de molécules LPS dans la membrane ou sur une modification des molécules LPS présentes, leur donnant une charge différente. En raison d’un changement de charge, l’attraction pour les polymyxines est réduite. Les molécules LPS se rapprochent également les unes des autres, ce qui rend plus difficile la liaison des polymyxines à ces molécules. Des recherches récentes ont montré que la résistance médiée par les plasmides peut être trouvée chez des souches d’E. coli isolées chez des animaux. Pour ce groupe d’antibiotiques, une résistance croisée complète se produit ; les bactéries résistantes à une polymyxine sont similaires aux autres polymyxines. Aux Pays-Bas, la résistance à la colistine est présente chez Salmonella spp. chez les bovins.
Les fluoroquinolones inhibent l’action de l’ADN gyrase et de la topoisomérase. L’une des fonctions de ces enzymes est le contrôle des brins d’ADN pour la prévention des mutations. L’inhibition de ces enzymes assure Ainsi, pour une augmentation du nombre de mutations dans l’ADN bactérien. Naturellement, cela augmente également la probabilité de mutations codant pour les mécanismes de résistance.
La résistance aux fluoroquinolones résulte d’une modification du site de liaison au niveau des enzymes, d’une perméabilité réduite, d’un pompage d’efflux et/ou d’un blindage du site de liaison. La perméabilité réduite est causée par une diminution du nombre de pores OMP (protéines de la membrane externe) dans la membrane externe de grammes de bactéries négatives. Si cette forme de résistance se produit, cela conduira généralement à une résistance à d’autres antibiotiques qui utilisent également ces pores tels que les tétracyclines et les antibiotiques β-lactamines.
Lorsque les bactéries développent une résistance à l’une des fluoroquinolones, elles sont généralement moins sensibles aux autres fluoroquinolones. Cela est particulièrement vrai pour les molécules plus anciennes et pour les bactéries qui ont de multiples mécanismes de résistance se sont développées. Aux Pays-Bas, les bactéries résistantes aux fluoroquinolones sont régulièrement isolées. Par exemple, la résistance à l’enrofloxacine est connue pour E. coli (boeuf) et Enterococcen spp. (volailles). La résistance à la fluméquine se produit chez les bovins (E.coli, Salmonella spp., M.Haemolytica et Pasteurella spp.), les poulets (E.coli) et les porcs (B.Bronchiseptica).
Diaminopyrimidines et sulfonamides
Pharmacodynamique
Les diaminopyrimidines et les sulfamides inhibent la synthèse de l’ADN des bactéries en inhibant la synthèse de l’acide folique. Les sulfamides interfèrent avec la première étape de la synthèse de l’acide folique, l’étape dans laquelle l’acide para-aminobenzoïque (PABA) est ajouté au diphosphate dihydroptéoate. Les sulfonamides sont des analogues structuraux du PABA et rivalisent pour l’enzyme dihydroptéroate synthétase.
Les diaminopyrimidines interviennent plus tard dans le processus et provoquent une inhibition compétitive de l’enzyme dihydrofolate réductase, provoquant empêche le dihydrofolate d’être converti en tétrahydrofolate, la forme active de l’acide folique.
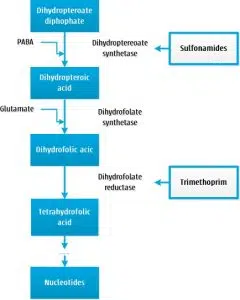
Les sulfamides sont bactériostatiques, les diaminopyrimidines sont bactéricides dépendants de la dose. La combinaison de ces antibiotiques est bactéricide. Comme expliqué plus haut, l’action des sulfamides est basée sur un lien concurrentiel avec PABA. Dans un environnement avec une forte concentration de PABA, les sulfamides ne remporteront pas cette compétition et par conséquent sera réduit l’efficacité. Des concentrations élevées de PABA se trouvent dans les inflammations avec beaucoup de pus, de nécrose et/ou de débris. Cela explique pourquoi les sulfamides in vivo peuvent parfois être insuffisamment efficaces malgré une bonne sensibilité in vitro.
Les diaminopyrimidines et les sulfamides ont un large spectre d’action. Cela peut être expliqué simplement parce que l’acide folique est essentiel à la production d’ADN et d’ARN chez presque toutes les bactéries. Seuls quelques Enterobactériaceae sont capables de dissoudre l’acide folique prendre de leur environnement, réduisant ainsi leur sensibilité aux sulfamides.
La synthèse de l’acide folique est également importante chez les mammifères, en particulier pendant la grossesse. Bien que la dihydrofolate réductase bactérienne soit beaucoup plus sensible au triméthoprime que la même enzyme utilisée chez les mammifères, ces antibiotiques doivent être utilisés avec prudence chez les animaux enceintes.
Constante de lipophilité et de dissociation
Les sulfamides et les diaminopyrimidines ont une lipophilité modérée. La liaison aux protéines plasmatiques des sulfamides est généralement élevée, mais cette liaison varie considérablement entre les différents sulfamides, mais aussi entre les espèces animales. Les diaminopyrimidines ont une liaison modérée aux protéines plasmatiques. Le volume de distribution des sulfamides est faible, mais ils se propagent généralement bien à travers les tissus, y compris les liquides synoviaux et céphalo-rachidiens. Les diaminopyrimidines ont un plus grand volume de distribution et, contrairement aux sulfamides, sont également intracellulaire.
Aussi le pKa des deux substances diffère : les sulfamides ont un pKa plus faible que les diaminopyrimidines. Cela provoque l’accumulation des sulfamides dans les tissus dont le pH est inférieur au pH plasmatique, tandis que les diaminopyrimidines s’accumulent dans les tissus à pH relativement élevé.
L’ élimination des sulfamides et des diaminopyrimidines se fait en partie par biotransformation dans le foie, suivie d’une excrétion par les reins, la bile, le lait et les fèces. En outre, une partie de ces antibiotiques est excrétée inchangée par les reins. Les différences de pH dans l’urine provoquent un taux d’élimination alternatif ; à un pH alcalin, une plus grande proportion de sulfamides sera excrétée inchangée alors qu’à un pH acide, une plus grande proportion de sulfamides sera excrétée inchangée. les diaminopyrimidines sont excrétées inchangées. Le taux de biotransformation est responsable des variations de demi-vie chez les sulfamides et les diaminopyrimidines à différentes les espèces animales.
En raison de la nature alcaline des sulfamides, l’injection de ces antibiotiques peut être irritant. Les sulfamides et le TMP/S doivent donc être administrés lentement.
Ratio et demi-vies
Lors de l’utilisation de produits combinés avec le triméthoprime et un sulfonamide, on essaie d’atteindre un rapport de 1:20 in vivo, puisque le triméthoprime est environ 20 fois plus puissant que les sulfamides. Il a été démontré que ce rapport est le mieux atteint en administrant un médicament contenant du triméthoprime et des sulfamides dans un rapport de 1:5.
Tableau 1 Demi-vie d’élimination du triméthoprime et de divers sulfamides chez les bovins, les veaux, les porcs et les chevaux après administration intraveineuse (h).
| Boeuf | Mollet | Cochon | Cheval | |
| Triméthoprime | 1,0 — 2,0 | 1,9 — 2,1 | 2,7 — 2,9 2* | 2,0 — 3.0* |
| Sulfadiazone | 2,5 | 4.4 1 | 2,8 | 4.6 |
| Sulfaméthoxazole | 2,3 | 12,8 * | 12,9 et 12,4 2 | 3,5 |
| Sulfachlorpyridazine | 1,2 | 13.1* | 3,0 4 | 3,8 |
| Sulfadoxine | 10,8 — 13,0 | 12.9 | 8,2 et 8,4 3 | 14.2 |
1 La demi-vie d’élimination chez les veaux passe de 5,7 à 3,6 entre 1 et 42 jours. 2 Demi-vie d’élimination chez les animaux sains et chez les animaux atteints de pneumonie, respectivement. Demi-vie d’élimination avec administration de sulfadoxine et de sulfadoxine avec triméthoprime, respectivement. Demi-vies d’élimination déterminées après administration orale de sulfachlorpyridazine chez les porcs. * La méthode d’administration n’est pas décrite dans l’étude.
Étant donné que la demi-vie d’élimination des sulfamides varie selon la substance active et entre les espèces animales, la La demi-vie du sulfonamide utilisé ne correspond pas toujours à la demi-vie du triméthoprime (voir tableau 1). Par conséquent, le rapport entre le triméthoprime et le sulfonamide ne correspondrait qu’à 1:20 pour une courte période de temps.
Malgré la différence de demi-vie, une bonne efficacité est généralement observée après l’administration de produits combinés avec le triméthoprime et les sulfamides. Il y a plusieurs facteurs qui peuvent jouer un rôle à cet égard. Premièrement, le grand volume de distribution du triméthoprime garantit que les concentrations dans les tissus sont plus élevées que les concentrations plasmatiques et sont maintenues sur une période plus longue. De plus, il a été démontré que la synergie se produit in vitro à une marge plus large que 1:20 (c.-à-d. 1:1 à 1:1000). Enfin, la pharmacocinétique chez un animal malade ou au site de l’inflammation peut être différente de celle des animaux en bonne santé.
Concentration ou effet dépendant du temps
Les deux Les sulfonamides sous forme de diaminopyrimidines ont un effet dépendant du temps. Cela signifie qu’il est important que la concentration de ces antibiotiques soit au moins égale à la CMI (concentration minimale inhibitrice) de la bactérie à traiter aussi longtemps que possible.
Combinaison d’antibiotiques
Les diaminopyrimidines et les sulfamides sont souvent combinés les uns avec les autres pour obtenir un effet bactéricide. De plus, dans certaines situations, la combinaison TMP/S peut également être combinée avec d’autres antibiotiques pour élargir le spectre d’action aux bactéries aérobies et anaérobies, même dans des situations où du pus ou des débris peuvent se produire. Cela peut être réalisé, entre autres choses, par une combinaison avec une pénicilline.
Il n’est pas recommandé de combiner TMP/S avec d’autres médicaments vétérinaires dans l’eau potable. La combinaison du triméthoprime et des sulfamides est difficile à soluble parce que le triméthoprime se dissout dans un environnement acide, tandis que les sulfamides se dissolvent dans un environnement de base. Les excipients peuvent augmenter la solubilité, mais cet effet peut être neutralisé par d’autres médicaments vétérinaires ou excipients ajoutés à d’autres médicaments vétérinaires.
En outre, les sulfamides ne doivent pas être combinés avec la procaine (ou la pénicilline de procaïne), car il s’agit d’un analogue de PABA et est en concurrence avec les sulfamides.
Résistance
La résistance aux sulfamides est régulièrement démontrée chez les bactéries isolées de bovins, de porcs et de volailles, en particulier E. coli et certaines espèces de Salmonella. La résistance aux sulfamides peut être liée à la fois chromosomique et plasmide et peut être causée par trois mécanismes :
- Changement de l’enzyme dihydroptéroate synthétase qui empêche les sulfonamides de se lier ;
- Réduction de la concentration intracellulaire due à une perméabilité réduite pour les sulfamides ;
- Production de PABA ce qui entraînera la perte de l’obligation concurrentielle des sulfamides.
Les bactéries résistantes à un sulfonamide sont également résistantes aux autres sulfamides (résistance croisée complète). La multirésistance est connue dans laquelle les bactéries sont également résistantes à d’autres antibiotiques tels que le triméthoprime ou la streptomycine. Ceci est particulièrement observé dans la résistance aux plasmides.
La résistance aux diaminopyrimidines est souvent basée sur un changement de l’enzyme dihydrofolate réductase qui empêche les antibiotiques de se lier à elle, mais aussi une perméabilité réduite se produit. Cette résistance peut également être liée à la fois chromosomique et plasmide.
Tétracyclines et aminoglycosides
Pharmacodynamique
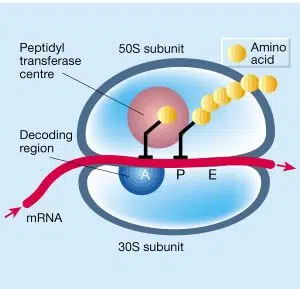
Les tétracyclines et les aminoglycosides entrent dans une liaison réversible avec la sous-unité 30S du ribosome. La sous-unité 30S du ribosome est la petite sous-unité, qui est responsable du décodage de l’ARN de sorte que le ribosome sait quels acides aminés devraient être intégrés dans la protéine.
Les tétracyclines interviennent dans la première étape de ce processus ; la liaison de l’ARNt au ribosome. L’ARN ne peut pas être lu et aucun acide aminé ne peut être ajouté à la chaîne protéique. Comme aucune protéine n’est formée, la bactérie ne peut pas se multiplier. Cela conduit à un effet bactériostatique.
Les aminoglycosides se lient à un autre récepteur de la sous-unité et s’assurent que l’ARN peut se lier mais qu’il est mal lu. Les mauvais acides aminés sont ensuite ajoutés à la chaîne protéique. Entre autres choses, les protéines mal composées sont intégrées dans la paroi cellulaire, ce qui peut entraîner une perturbation de la perméabilité et finalement une lyse de la bactérie. Ceci explique pourquoi les aminoglycosides, contrairement à d’autres inhibiteurs de la synthèse des protéines, sont bactéricides.
Constante de lipophilité et de dissociation
Dans l’introduction, déjà décrit que la lipophilité et le pKa sont importants pour le passage des membranes. Pour les antibiotiques discutés ici, cela est très important car ils doivent passer à travers la membrane cellulaire pour atteindre le ribosome.
Les tétracyclines ont une lipophilité modérée en combinaison et un grand volume de distribution. L’absorption du tractus gastro-intestinal est raisonnable, mais les tétracyclines sont des substances chélatantes qui se lient à la nourriture. Par conséquent, la présence de nourriture dans le tractus gastro-intestinal affecte négativement son absorption. En raison du grand volume de distribution, les concentrations tissulaires sont généralement plus élevées que les concentrations plasmatiques. Les tétracyclines atteignent des concentrations particulièrement bonnes dans les poumons et les mamelles, mais dans le liquide céphalo-rachidien, les concentrations actives sont rarement atteintes en raison d’une lipophilité modérée.
La doxycycline a une lipophilité plus élevée que les autres tétracyclines, ce qui améliore l’absorption par le tractus gastro-intestinal et atteint des concentrations plus élevées se trouvent dans les tissus. Les aminoglycosides ont une faible lipophilité et un faible volume de distribution. Cela signifie que la biodisponibilité orale est mauvaise et que les aminoglycosides administrés par voie orale sont particulièrement efficaces dans l’intestin. Après administration parentérale, la distribution est principalement déterminée par la liaison aux protéines plasmatiques ; les antibiotiques liés aux protéines plasmatiques ne quittent pas les vaisseaux sanguins et sont donc inefficaces. Étant donné que les aminoglycosides ont une faible liaison aux protéines plasmatiques, ils atteignent des concentrations efficaces dans les liquides péricardiques, synoviaux, pleuraux, périlymphatiques et péritonéaux après administration parentérale. La diffusion vers les articulations s’améliore quand elles sont enflammées, car le pH dans les articulations diminue et le piégeage ionique se produit en raison de la forte pKa des aminoglycosides. Les aminoglycosides atteignent le liquide céphalo-rachidien, ainsi que les tétracyclines, à des concentrations insuffisamment élevées pour être efficaces.
Dans les tétracyclines, le passage au-dessus du membranes cellulaires se produisent en raison de la diffusion, mais les aminoglycosides sont activement pompés dans la cellule par gramme de bactéries négatives par un processus dépendant de l’oxygène. Cela explique pourquoi ces antibiotiques sont inefficaces dans des conditions anaérobies et dans des bactéries anaérobies (facultatives). En outre, les aminoglycosides sont réduits en travaillant dans un environnement avec des débris purulents élevés et de faibles valeurs de pH, comme cela peut se produire avec des dommages tissulaires. Les débris contiennent de nombreuses protéines, qui peuvent se lier aux aminoglycosides, de sorte qu’ils ne peuvent plus passer à travers la membrane cellulaire de la bactérie et sont donc inefficaces. Bien que le drainage soit souvent effectué pour d’autres raisons, le drainage dans une inflammation purulente peut contribuer à une amélioration de l’efficacité de l’antibiotique.
Concentration ou effet dépendant du temps
Les aminoglycosides ont une efficacité dépendante de la concentration et un long effet post-antibiotique. Il est donc important d’avoir un dosage suffisamment élevé au site de l’infection, mais en raison de l’effet post-antibiotique long, il n’est pas nécessaire d’atteindre en permanence des concentrations supérieures à la CMI. En outre, l’administration répétée d’aminoglycosides en une journée est un facteur de risque important pour l’apparition de la néphrotoxicité. Les aminoglycosides sont excrétés par voie rénale et s’accumulent dans les cellules tubulaires, où ils peuvent causer une toxicité. L’accumulation, et donc la toxicité, dépend en particulier de la durée de l’exposition et est minime à une concentration maximale courte. La combinaison de ces deux facteurs conduit au fait que les aminoglycosides sont de plus en plus administrés qu’une fois par jour.
L’ accumulation rénale est également la cause de la longue période d’attente à suivre après l’utilisation des aminoglycosides. Les tétracyclines n’ont pas d’effets spécifiques liés à la concentration ou au temps. Tant la concentration que la durée de l’exposition sont de intérêt.
Combinaison d’antibiotiques
Dans certaines situations, il peut être utile de combiner des antibiotiques, par exemple pour élargir le spectre. Cependant, les inhibiteurs de synthèse protéique ne peuvent pas être combinés avec des antibiotiques β-lactamines, tels que les pénicillines. L’efficacité des antibiotiques β-lactamines dépend du partage des bactéries, ce qui ne se produira pas après l’administration d’inhibiteurs de synthèse protéique. Cependant, les aminoglycosides sont une exception à cela. En raison de l’utilisation d’antibiotiques β-lactamines, la paroi devient plus perméable aux aminoglycosides. En outre, les aminoglycosides, étant le seul dans le groupe des inhibiteurs de la synthèse des protéines, sont bactéricides. Cette combinaison peut entraîner un effet synergique contre Streptococcen spp., Enterococcen spp., Pseudomonas spp. et d’autres grammes de bactéries négatives.
Résistance
En raison de l’utilisation répandue des tétracyclines, la résistance est observée chez les bovins, les porcs et les volailles. Là sont plusieurs mécanismes sur lesquels la résistance aux tétracyclines peut se développer :
- Changement ou blindage du point d’engagement sur le ribosome
- Réduction de la concentration intracellulaire de l’antibiotique
- Ceci peut être réalisé par des pompes à efflux ; ces pompes dépendantes de l’énergie échangent un proton contre un complexe d’une tétracycline avec un cation.
- Il peut également y avoir une diminution de la production de porine OPPF. C’est la structure à travers laquelle les tétracyclines passent à travers la membrane externe de la bactérie.
- Inactivation enzymatique des antibiotiques (en particulier chez les Enterobactériaceae)
Le changement de ribosome et l’utilisation de pompes à efflux sont les principaux mécanismes. La résistance croisée fait partie de ce groupe d’antibiotiques règle ; la résistance à un antibiotique de ce groupe conduit également à une résistance à d’autres tétracyclines. Aussi contre les aminoglycosides il y a dans le pratiquer la résistance. La résistance aux aminoglycosides est principalement obtenue par inactivation enzymatique de l’antibiotique, mais les autres mécanismes de résistance mentionnés ci-dessus pour les tétracyclines sont également connus pour les aminoglycosides. La résistance croisée aux aminoglycosides est très imprévisible ; dans de nombreux cas, la bactérie est toujours sensible à un autre aminoglycoside, mais elle doit être testée au préalable.
Macrolides, lincosamides, pleuromutilines et phénicols
Pharmacodynamique
Les antibiotiques des groupes de macrolides, lincosamides, pleuromutilinines et phénicols se lient à la sous-unité 50S du ribosome. La sous-unité 50S est la grande sous-unité et est responsable de la fusion des divers acides aminés pour former une seule chaîne (peptide). Cela dépend en particulier de la peptidyl transférase e nzym.
Les lincosamides, les pleuromutilines et les phénicols inhibent l’action de l’enzyme peptidyltransférase susmentionnée. Pour cette raison, les acides aminés ne sont pas fusionnés en une seule chaîne (peptide) avant de quitter le ribosome. Les macrolides ont un mécanisme d’action légèrement différent ; ils assurent que la chaîne protéique formée est libérée avant qu’elle ne soit terminée. Ainsi, en présence de macrolides, seules des chaînes protéiques courtes et incomplètes sont formées, alors qu’en présence d’un antibiotique de l’un des autres groupes, les chaînes protéiques ne se forment pas du tout.
Ces antibiotiques sont généralement classés comme bactériostatiques. Cependant, dans certains cas, l’effet est bactéricide. Cela dépend de la concentration de l’antibiotique, de la période pendant laquelle la concentration est supérieure à la CMI, de la souche bactérienne traitée et de la quantité de bactéries. Pour le florfénicol, un effet bactéricide a été démontré contre Actinobacillus pleuropneumoniae et Pasteurella multocida lorsque la concentration de florfénicol pendant 12 heures était plus élevé que le MIC.
En plus de l’effet antibactérien bactériostatique, les macrolides et les lincosamides sont parfois appelés médicaments vétérinaires immunomodulateurs. Il existe plusieurs façons dont ces antibiotiques ont un effet sur l’évolution de l’infection ; ils affectent à la fois la bactérie et l’interaction entre la bactérie et l’hôte ainsi que le système immunitaire de l’hôte. Cependant, ces effets ne sont obtenus que par les macrolides qui possèdent un cycle lactone vide de 14 ou 15. Parmi ceux-ci, seule l’érythromycine est appliquée vétérinaire. En outre, afin d’obtenir cet effet, ces antibiotiques doivent être administrés à une faible concentration pendant une longue période de temps, ce qui ne correspond pas dans le cadre d’une utilisation responsable des antibiotiques.
De plus, des études ont montré que la tilmicosine est également efficace dans les infections à SPRR. Les études in vitro décrivent une diminution de la concentration virale dans les lignées cellulaires, alors qu’in vivo montrent une diminution de la mortalité, le nombre de lésions pulmonaires et les titres viraux dans les poumons et le sérum. Cet effet s’explique peut-être par l’accumulation de tilmicosine dans les macrophages, où ils empêchent la propagation du virus.
Constante de lipophilité et de dissociation
Les membranes de passage est très important pour ce groupe d’antibiotiques, puisque le point d’application (ribosome) est dans la bactérie. Comme décrit précédemment, la lipophilité et le pKa sont importants pour cela. Les antibiotiques qui se lient à la sous-unité 50S du ribosome ont une lipophilité modérée à élevée avec un pKa élevé. En raison d’une bonne lipophilité, ils sont capables de passer des membranes biologiques, y compris la membrane cellulaire. En raison de la forte pKa de ces antibiotiques, le piégeage ionique se produit dans les organes dont le pH est inférieur au pH du plasma (p. ex. poumons, pis). Aussi le piégeage ionique se produira dans les tissus enflammés, car ici aussi le pH du tissu est inférieur à le pH du plasma.
En raison du grand volume de distribution de ces antibiotiques, des concentrations supérieures aux concentrations sériques seront souvent atteintes dans les tissus. La biodisponibilité est bonne après administration orale et parentérale. La tulathromycine est un macrolide semi-synthétique avec une pharmacocinétique spécifique. La recherche a montré que cet antibiotique atteint une concentration élevée et homogène dans les poumons chez les porcs après administration intraveineuse ou intramusculaire, qui est en outre conservée pendant une longue période de temps. Il a également été démontré que la tulathromcyine possède des propriétés pharmacocinétiques similaires chez d’autres espèces animales telles que les bovins, les chevaux et les chèvres.
Concentration ou effet dépendant du temps
Les macrolides, les lincosamides, les pleuromutilines et les phénicols sont des antibiotiques dépendants du temps. Il est donc important de maintenir les concentrations sur le site de l’infection pendant une période suffisamment longue. qui sont au moins aussi élevés que le MIC. Le dosage continu est donc préférable au dosage par impulsion.
Combinaison d’antibiotiques
Il n’est pas recommandé de combiner les antibiotiques de ce groupe avec des antibiotiques β-lactamines. L’effet de ce groupe d’antibiotiques est décrit comme bactériostatique et bactéricide, mais l’effet sera certainement bactériostatique pendant une partie du temps. La croissance des bactéries est alors inhibée, ce qui entraîne la perte d’efficacité des antibiotiques β-lactamines car ils ne sont efficaces que contre la division des bactéries.
En outre, lors de la combinaison de plusieurs antibiotiques de ce groupe, il faut être prudent. Les macrolides, les lincosamides, les pleuromutilinines et les phénicols doivent pouvoir se lier à la sous-unité 50S pour être efficaces. Les sites de liaison des macrolides, des lincosamides et des phénicols se chevauchent partiellement. Combiner ces antibiotiques les uns avec les autres est donc non recommandé.
Une combinaison avec les tétracyclines ou les aminoglycosides est possible parce qu’ils se lient à l’autre sous-unité du ribosome.
Enfin, lors de la combinaison de ces antibiotiques, il est nécessaire de prendre en compte le fait qu’ils inhibent le système enzymatique CYP-450 (cytochrome P450) dans le foie. L’inhibition de ces enzymes par des antibiotiques entraîne le métabolisation de l’antibiotique lui-même, ainsi que d’autres médicaments vétérinaires (fluoroquinolones, ionophores). Un surdosage relatif peut alors en résulter.
Résistance
La résistance aux macrolides, aux pleuromutilinines et aux lincosamides chez les bactéries Gram négatif est généralement basée sur une perméabilité à la membrane cellulaire réduite. Dans les bactéries Gram positif, la résistance à ces antibiotiques est souvent causée par un changement dans le point d’application, l’efflux actif des antibiotiques et l’inactivation enzymatique des antibiotiques. Ces mécanismes de résistance se produisent également dans Moins fréquent chez les bactéries Gram négatif.
Lorsque la résistance est causée par la modification du point d’application, la résistance croisée se produit souvent entre les macrolides, les lincosamides et la streptogramine B. Ceci s’explique par le chevauchement des sites de liaison de ces antibiotiques. Ce mécanisme de résistance est également connu sous le nom de résistance MLSB. La résistance aux phénicols dans la plupart des cas est basée sur l’inactivation enzymatique de l’antibiotique. Cependant, les systèmes d’efflux sont également connus. Les bactéries Gram négatif trouvent aussi parfois une perméabilité réduite ou un transporteur efficace contre divers antibiotiques (bactéries multirésistantes).
Références
- Altenburg, J., de Graaf, C.S., van der Werf, T.S., Boersma, W.G. (2011) Effets immunomodulateurs des antibiotiques macrolides — Partie 1 : Mécanismes biologiques. Respiration, 81:67 -74.
- Baert, K., de Baere, S., Croubels, S., Gasthuys, F., de Backer, p. (2001) Pharmacocinétique et biodisponibilité de la sulfadiazine et du triméthoprime (trimazine 30%) après administration orale chez les jeunes porcs non à jeun. J Vet Pharmacol Therap 24:295-298.
- Barberio, A., Badan, M., Bonamico, S., Mancin, M., Simonato, G., Paroline, O. Bazzim, D. (2012) Utilisation du sulfate d’aminosidine pour prévenir la cryptosprodiose chez les veaux. Examen de la médecine vétérinaire 47.
- Benchaoui, H.A., Nowakowski, M., Sherington, J, Rowan, T.G., Sunderland, S.J. (2004) Pharmacocinétique et concentrations de tulathromycine dans les tissus pulmonaires chez les porcs. J.Vet.Pharmacol.Therap 27:203-210.
- Bleyen, N., de Gussem, K., Nguyen, A.D., Ons, E., van Gerven, N. Goddeeris, B. (2009) Effets non curatifs, mais prophylactiques de la paromomycine chez les dindes infectées par Histomonas meleagridis et son effet sur la performance chez les dindes non infectées. Document de recherche, Université de Louvain, Belgique.
- Brouwers, J.R.B.J. (1987) Pharmacocinétique des fluoroquinolones plus récentes. Pharmaceutisch Weekblad Scientifique Edition (Supplément) 9 : S16-S22.
- Brown, M.P., Gronwall, R., Castro, L. (1988) Pharmacocinétique et concentrations de triméthoprim-sulfaméthoxazole dans les juments dans les liquides corporels et endométriaux. Am J Vet Res 49 : 918-922.
- Centre pour la sécurité alimentaire et la santé publique — Université d’État de l’Iowa (2011) Staphylococcus aureus résistant à la méthicilline.
- Fiche d’information du Centre pour la sécurité alimentaire et la santé publique (2011) Staphylococcus aureus résistant à la méthicilline.
- Du, Y., Yoo, D., Paradis, M-A, Scherba, G. (2011) Activité antivirale de la tilmicosine pour le virus du syndrome reproducteur et respiratoire porcin de type 1 et de type 2 dans les macrophages alvéolaires porcins cultivés. Antivir Antirétrovir 3 : 28-33.
- Formuarlium melkvee, werkgroep veterinair antibioticumbeleid KNMVD. Versie 1.1 (2012), aangepast op 02-01-2014.
- Giguere, S., Prescott, J.F., Baggot, J.D., Walker, R.D., Dowling, P.M. (2007) La thérapie antimicrobienne en médecine vétérinaire, quatrième édition. Blackwell édition, 121-137, 179 — 188, 191 — 205, 207 — 229, 231-262.
- Guardabassi, L., Jensen, L.B., en Kruse, H. (2008) Guide de l’utilisation des antimicrobiens chez les animaux. Blackwell Publishing.
- Gustafsson, A., Baverud, V, Franklin, A., Gunnarsson, A., Ögren, G., Ingvast-Larsson, C. (1999) Administration répétée de triméthoprim/sulfadiazine dans la pharmacocinétique du cheval, liaison aux protéines plasmatiques et influence sur la microflore intestinale. J Vet Pharmacol Therap 22:20-26.
- Hamilton-Miller, J.M.T. en Shah, S. (1999) Effet de la concentration d’antibiotiques sur la mort de Staphylococcus aureus et d’Enterococcus faecalis : Comparaison du nouveau penem, Men 10700, avec d’autres antibiotiques β-lactamiques. J Antimicrobes chimiautre 44 : 418-420.
- He, J., Tang, S., Li, L., Zhang, C., Li, X., Xia, Xia, X., Xiao, X. (2010) Pharmacocinétique d’une nouvelle suspension d’amoxicilline/colistine après administration intramusculaire chez des porcs. J vétérinaire Pharmacol Therap 34:42-50.
- Rapport scientifique conjoint de l’ECDC, de l’EFSA et de l’EMEA sur le Staphylococcus aureus résistant à la méthicilline (SARM) chez le bétail, les animaux de compagnie et les aliments.
- Kaartinen, L., Gips, M., Laurila, T., Härtel, H., Soback, S., Pyörälä (2000) Pharmacocinétique de la sulfadoxine et du triméthoprime et irritation des tissus causée par deux produits contenant du sulfate triméthoprime après administration sous-cutanée chez des veaux préruminants. Ré. vétérinaire 31 : 517-526.
- Kaartinen, L., Löhönen, K., Wiese, B., Franklin, A., Pyörälä, S (1999) Pharmacocinétique de sulfadiazine-triméthoprime chez les vaches laitières allaitantes (résumé). Acta Vet Scand 40:271-278.
- Kwiatkowska, B., Maslinska, M., Pryzgodzka, M., Dmowksa-Chalaba, J., Dabrowska, J., Sikorska-Siudek, K. (2013) Système immunitaire comme nouvelle cible thérapeutique pour les antibiotiques. Progrès dans les domaines des biosciences et de la biotechnologie, 4:91-101.
- Leclercq, R., Courvalin, P. (2002) Résistance aux macrolides et aux antibiotiques apparentés chez Streptococcus pneumonie.Agents antimicrobiens et chimiothérapie 46:2727-2734.
- Melchior, M., Van Hout-van Dijk, J. (2011) Antibiotica, van werkingsmechanismen naar antibactériële therapie. Deel III (fourgon IV). Tijdschrift voor Diergeneeskunde 136 (9) : 646-652.
- Mengelers, M.J.B., van Gogh, E.R., Kuiper, H.A. Pijpers, A., Verheijden, J.H.M., van Miert, A.S.J.P.A.M. (1995) Pharmacocinétique de la sulfadiméthoxine et du sulfaméthoxazole en association avec le triméthoprime après administration intraveineuse à des porcs sains et pneumoniques. J Vet Pharmacol Therap 19:243-253.
- Surveillance jaarverslag 2012, Gezondheidsdienst voor dieren te Deventer.
- Nielsen, P., Rasmussen, F. (1975) Triméthoprime et suladoxine chez les porcs ; Demi-vies, volume de distribution et concentration tissulaire. Zbl Vet Med A 22 : 564-571.
- Nielsen, P., Rasmussen, F. (1977) Demi-vie, volume apparent de distribution et liaison aux protéines pour certains sulfamides chez les vaches. Recherche en sciences vétérinaires 22 : 205-208.
- Nielsen, P., Romvary, A., Rasmussen, F. (1978) Sullphadoxine et triméthoprime chez les caprins et les vaches : fraction d’absorption, demi-vie et effet dégradant de la flore ruminale. J Vet Pharmacol Therap 1:37-46.
- Openstax CNX, Diversité probiotique. (bron voor afbeelding).
- Pankey, G.A., Sabath, L.D. (2004) Pertinence clinique du mécanisme d’action bactériostatique par rapport au mécanisme d’action bactéricide dans le traitement des infections bactériennes à Gram positif. Maladies infectieuses cliniques 38 ; pp. 864-870.
- Plumb, D.C. (2011) Manuel des médicaments vétérinaires de Plumb, 7eédition. Wiley-Blackwell.
- Quintiliani, R. Pharmacodynamique des agents antimicrobiens : tuerie dépendante du temps par rapport à la concentration.
- Rasmussen, F., Gelsa, H., Nielsen, P., Pharmacocinétique de la sulfadoxine et du triméthoprime chez les chevaux. Demi-vie et volume de distribution de sulfadoxine et triméthoprime et excrétion cumulative du -triméthoprime. J Vet Pharmacol Therap 2:245-255.
- Rolinski, Z., Duda, M. (1984) Pharmacocinétique analyse du niveau de combinaison sulfamide-triméthoprime chez les veaux (abstrait). Pol J Pharm Pharm 36 : 35-40.
- Romvary, A., Horvay, A. (1976) Données sur la pharmacocinétique des combinaisons sulfonamide-triméthoprime chez les porcs suceurs. Zbl Vet Med 23:781-792.
- Schwarz, S. Chaslus-Dancla, E. (2001) Utilisation d’antimicrobiens en médecine vétérinaire et mécanismes de résistance. Vet Res 32 : 201-225.
- Shoaf, S.E., Schwark, W.S., Guard, C.L. (1989) Pharmacocinétique de la sulfadiazine/triméthoprime chez les veaux mâles néonataux : effet de l’âge et de la pénétration dans le liquide céphalo-rachidien (résumé). Am J Vet Res 50 : 396-403.
- Shoaf, S.E., Schwark, W.S., Guard, C.L., Swartsman, R.V. (1986) Pharmacocinétique de la triméthoprim/sulfadiazine chez les veaux néonatals : influence de la synovite. J Vet Pharmacol Therap 9:446-454.
- van Duijkeren, E., Vulto, A.G., Sloet van Oldruitenborgh, M.M., Kessels, B.G., van Miert, A.S., Breukink, H.J. (1995) Pharmacocinétique de triméthoprime/ sulfachlorpyridazine chez les chevaux après administration orale, nasogastrique et intraveineuse. J Vet Pharmacol Ter 18:47 -53.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie I (de la IV). Journal of Veterinary Medicine 136:494 -499.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie II (de la IV). Journal of Veterinary Medicine 136 (8) : 572-577.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie III (de la IV). Journal de médecine vétérinaire 136:646 -652.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie Lv (de IV). Journal of Veterinary Medicine 136 (10) : 730-733.
- Villarino, N., Brown, S.A., Martin-Jiménez (2013) Comprendre la pharmacocinétique de la tulathromycine : une perspective pulmonaire. Vétérinaire. Pharmacol. Therap. 37:211-221.
- Villarino, N., Lesman, S., Fielder, A., Garciá-Tapia, D., Cox, S., Lucas, M., Robinson, J., Brown, S.A., Martin-Jiménez, T. (2012a) Pharmacocinétique pulmonaire de la tulathromycine chez le porc. Partie I : L’homogénat pulmonaire chez les porcs et les porcs sains a été confronté intratrachéalement au lipopolysaccharide d’Escherichia coli. Vétérinaire. Pharmacol. Therap 36:329-339.
- Villarino, N., Lesman, S., Fielder, A., Garciá-Tapia, D., Cox, S., Lucas, M., Robinson, J., Brown, S.A., Martin-Jiménez, T. (2012b) Pharmacocinétique pulmonaire de la tulathromycine chez le porc. Partie 2 : Compartiments intra-voies respiratoires J. Vet. Pharmacol. Therap 36:340-349.
- Viu, M., Quilez, J., Sanchez-Acedo, C., del Cacho, E., Lopez-Bernad, F. (2000) Essai sur le terrain sur l’efficacité thérapeutique de la paromomycine sur l’infection naturelle à Cryptosporidium parvum dans les labmes. Zoot. Vétérinaire 28 : 13-19.
- Vologodskii, A. (2004) Analyse computationnelle de l’action de l’ADN gyrase. Biophysique journal 87 : 3066-3073.
- Williamson, J.R. (2000) Petite sous-unité, grande science. Biologie moléculaire. Nature 407:306 -307.